Tadalafil entfaltet seine Wirkung über eine selektive Hemmung der PDE5, wodurch die Konzentration von cGMP im glatten Muskelgewebe stabil bleibt. Diese biochemische Modulation resultiert in einer langanhaltenden Relaxation der Gefäßwände. Der Wirkstoff wird nach oraler Einnahme effizient resorbiert, mit einer Bioverfügbarkeit von rund 80 %. Seine Halbwertszeit von bis zu 36 Stunden ist innerhalb dieser Substanzklasse außergewöhnlich. Abgebaut wird er in der Leber, hauptsächlich durch CYP3A4, mit anschließender biliärer Exkretion. Typische unerwünschte Wirkungen entstehen durch eine verstärkte Vasodilatation, etwa Kopfschmerzen oder Flush. Pharmakologisch wird cialis generika vor allem durch die verlängerte Wirkungsdauer charakterisiert.
8. nikisch###_umbruchvorlage
EU RO PE AN JOUR NAL OF MED I CAL RE SEARCH
Eur J Med Res (2008) 13: 579-584 I. Holzapfel Publishers 2008
THREE-YEAR FOLLOW-UP OF A PATIENT WITH EARLY-ONSET
ALZHEIMER'S DISEASE WITH PRESENILIN-2 N141I MUTATION
G. Nikisch1, A. Hertel2, B. Kießling1, Th. Wagner1, D. Krasz1, E. Hofmann3, G. Wiedemann1
1Department of Psychiatry and Psychotherapy (Chief of Department: Prof. Dr. med. Georg Wiedemann ),
2Department of Nuclear Medicine, (Chief of Department: Assistant Prof. Dr. med. Andreas Hertel),
3Department of Neuroradiology, (Chief of Department: Prof. Dr. med. Erich Hofmann),
Abstract
early onset (onset < 65 years) demonstrate a faster pro-
Autosomal dominant early-onset Alzheimer disease
gression and a more severe clinical presentation than
(EOAD) is a heterogeneous condition that has been
those with a late onset (onset > 65 years), despite being
associated with mutations in 3 different genes: the
younger. Autosomal dominant early-onset Alzheimer
amyloid precursor protein (APP), presenilin 1
disease (EOAD) is a heterogeneous condition caused
(PSEN1), and presenilin 2 (PSEN2) genes. Most cases
by different genetic defects. Familial studies, in early-
are due to mutations in the PSEN1 gene, whereas mu-
onset cases, indicate molecular heterogeneity and have
tations in the APP and PSEN2 genes are rare. Muta-
linked EOAD to mutations in at least three genes: the
tion analysis of the APP, PSEN1 and PSEN2 genes
amyloid precursor protein gene (APP) located on chro-
was performed. We herein report the case of a Ger-
mosome 21 (Mullan and Crawford 1993) and the pre-
man EOAD patient with a family history of dementia
senilin 1 and 2 (PSEN-1 and PSEN-2, respectively)
and a missense mutation at codon 141 (N141I) of the
genes located on chromosome 14 and 1 (Levy-Lahad et
PSEN2 gene. To our knowledge, this is the first Ger-
al. 1995; Sherrington et al. 1995), respectively. Muta-
man EOAD patient without a Volga-German ancestry
tions in these genes account for approximately half of
and a positive family history for dementia carries the
the EOAD (Rosenberg 2000). Although more than 166
mutation PSEN-2 N141I. The patient came to our
mutations in the PSEN1 gene have been associated
clinic for the first time when she was 47 years old.
with autosomal dominant EOAD, only 13 such muta-
During the following 3 years, her Mini-Mental State
tions have been found in the PSEN2 gene (Finckh et
Examination (MMSE) score dropped from 28 to 0.
al. 2005). Mutations in the PSEN2 gene on chromo-
Mild cognitive impairment (MCI) was an early symp-
some 1 are the second most frequent form of familial
tom that was already present during the first consulta-
EOAD (age at onset 45-65 years) and have mainly been
tion. The concentration in cerebrospinal fluid (CSF)
described in large kindreds of Volga-Germans in the
of tau-protein (1151 pg/ml) was increased, whereas
U.S. (Levy-Lahad et al. 1995; Mann et al. 1997).
the concentration of beta-amyloid protein (Aß1-42)
It has been reported that 10-15% of patients with
was decreased (335 pg/ml). Magnetic resonance imag-
mild cognitive impairment (MCI) develop Alzheimer’s
ing (MRI) revealed only slight changes in the early
disease (AD) within 1 year (Peterson et al. 1999). Evi-
stage of the disease and positron emission tomogra-
dence shows that these pathological changes are de-
phy with [18F] fluoro-2-deoxy-D-glucose (18F-FDG
tectable before the onset of clinical dementia (Morris
PET) demonstrated glucose reduction left parietal and
and Price 2001). As clinically mildly demented AD pa-
in the precuneus region. Follow-up MRI and 18F-
tients show elevated tau-protein levels (Galasko 1998;
FDG PET studies showed progression of atrophy of
Riemenschneider et al. 1996) and decreased Aß1-42
the left entorhinal cortex with relative sparing of the
levels (Andreasen et al. 1999; Galasko 1998; Motter et
hippocampus and progressive hypometabolism of
al. 1995) in cerebrospinal fluid (CSF) compared to
both temporoparietal lobes and left frontal lobe.
controls. CSF tau protein and Aß1-42 have been pro-
posed as putative early diagnostic markers in MCI sub-
Key words: MCI; early-onset Alzheimer disease; prese-
jects. Patients who converted from MCI to AD
showed significantly higher tau-protein levels at base-
line compared to healthy individuals (Arai et al. 1997).
Moreover, it has been demonstrated that subjects with
MCI who later developed AD were identified by the
Alzheimer disease (AD) is characterized by memory
combination of decreased CSF concentrations of
loss and declining cognitive functions. Patients with an
Aß1-42 and increased levels of tau-protein (Andreasen
et al. 1999; Riemenschneider et al. 2002). These find-
frontotemporal theta wave activity. The EEG at the
ings suggest that tau-protein and Aß1-42 in CSF may
age of 51 years showed delta-theta wave activity with
be valuable to detect the preclinical stages of AD.
paroxysmal activation of frontal sharp and slow waves.
Up to now only two cases of EOAD due to muta-
tions in the PSEN2 gene have been identified in Ger-
many (Finckh et al. 2005). We describe the clinical
course of a patient with the N141I mutation of
The analysis of CSF revealed normal cell count and
protein content. There were no oligoclonal bands.
NSE and protein 14-3-3 were within the normal range.
Tau-protein was elevated 1151 pg/ml (normal range:
The patient was admitted to our psychiatric clinic for
47-225 pg/ml) and Aß1-42 decreased 335 pg/ml (nor-
the first time at the age of 48 years. From the age of
47 years she had started to complain about deficits in
her short-term memory. There was no previous histo-
ry of severe or chronic illnesses. The patient had no
evidence or history of neurological signs or symptoms
Our patient received a standardized clinical MRI scan
of cerebrovascular disease. Her orientation in person,
protocol with T1- weighted and T2- weighted se-
situation and time were adequate. She was unable to
quences to visualized structural brain changes as vol-
date biographical events of the few years and she was
ume reductions (i.e., atrophy). A PET scan, which im-
aware of her cognitive deficits. Physical examination
ages regional brain metabolism with the use of 18F-
and laboratory test was normal (including Vitamin
FDG, was obtained. At the first presentation MRI
B12, TSH, and TPHA). The patient had completed
scanning showed slightly changes at the age of 48 and
secondary school and education as an industrial clerk.
18F-FDG PET images already showed reduction of
She married at the age of 18, and gave birth to one
cortical glucose metabolism in the left parietal and
precuneus cortex (Fig. 2A). Follow-up MRI and 18F-
The family history revealed that the father (II-2)
FDG PET studies showed progression of atrophy of
and the grandmother (I-1) of the index patient suf-
the left entorhinal cortex with relative sparing of the
fered from AD (Fig. 1A). The grandmother died at age
hippocampus and progressive hypometabolism of bi-
52 years after a clinical course of approximately 5
laterally temporoparietal lobes with additional frontal
years on the sequelae of pneumonia with an onset at
hypometabolism more apperent on the left lobe three
48 years. The index patient’s father showed first mild
cognitive impairments at age of 64 years and died at
age 76 years. The 77-year old mother (II-1) of the in-
dex patient, and her three siblings aged 52, 50 and 46
(III-1, 3, 4, respectively) are asymptomatic at the time
After gaining written informed consent, a blood sam-
of clinical examination and genetic data were not col-
ple for genetic analysis was obtained from the
lected. The healthy son (IV-1) of the index patient re-
proband. The DNA was isolated according to stan-
ported here has a 50% chance of being a carrier of
dard procedures. The coding regions of the PSEN1,
the N141I mutation. Predictive testing was not per-
PSEN2, and APP (exons 2, 16 and 17) were amplified
using specific primers, as described previously (Cruts
et al. 1998; Mullan et al.) A heterozygote missense mu-
tation resulting in a substitution of asparagine at posi-
tion 141 by isoleucine (N141I) in exon 5 of PSEN2
The Mini-Mental-State-Examination (MMSE), the
Hamburg-Wechsler-Intelligence Test for Adults
(HAWIE) and the Hamilton Depression Scale (HAM-
The patient showed progressive mental decline. Neu-
D) were used to determine the patient’s cognitive abili-
ropsychological assessment, approximately 2 years af-
ties. At the first presentation the patient reached a
ter disease onset, shows marked intellectual loss. All
score of 28 points on the MMSE. The HAWIE
measures of auditory and visual memory for immedi-
showed a total IQ of 93 with a relevant discrepancy
ate and delayed recall were severely impaired (below
between the verbal IQ of 101 and the performance IQ
the first percentile). Acetylcholinesterase inhibitor and
of 84. This is mainly due to the inability of the patient
a NMDA receptor antagonist were prescribed
to distinguish between important and less important
(donepezil, followed by memantine) but were not ben-
information of the serial picture stories used in these
eficial; one year after presentation MMSE had
IQ subtests. The HAM-D showed a mildly elevated
dropped to 14 and to 0 two years later. The neuropsy-
chological profile was consistent with AD. When the
patient was 50 years old she was no longer able to take
care of herself. She became dependent with activities
such as shopping and housekeeping. At the age of 51
At the first presentation the electroencephalogram
years, the patient was unable to repeat words or to fol-
(EEG) at the age of 48 years showed a slow alpha
low instructions. She spoke only single words which
rhythm (6/s) with paroxysmal activation of irregular
were remotely connected to the actual situation.
to cosegregate in an autosomal dominant way with
EOAD in Volga-Germans in the U.S. (Levy-Lahad et
This case is unique in many respects. Although the
symptoms, signs, investigations, progress, and family
history are consistent with familial EOAD, the neu-
ropsychological follow-up changes of her affected rel-
atives – who shared a similar presentation – showed
the changes seen most often in MCI (Peterson et al.
1999). Cognitive deficits became obvious when she
was 47 years old with an onset of dementia in her
family at 48 years and 64 years. MCI was an early
symptom that was already present during the first con-
sultation. Our patient’s genetic analysis showed a het-
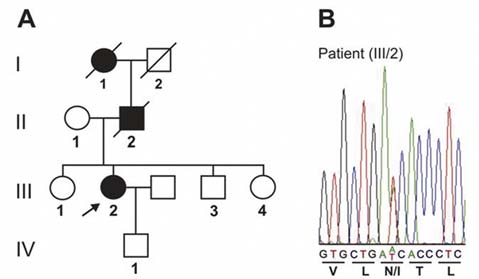
Fig. 1. (A): Pedigree of the German FAD family carrying the
erozygote missense mutation resulting in a substitu-
N141I mutation in PSEN2. Black symbols indicate affected
tion of asparagine at position 141 by isoleucine in
probands. The index patient is marked by an arrow. (B):
DNA sequence chromatogram of part of PSEN2 exon 5,
Although we were not able to analyse the PSEN-2
showing the heterozygous A to T transversion in the index
gene in the other two affected relatives, the detected
patient (III-2). Corresponding reading frame and amino acid
PSEN-2 mutation strongly suggests a genetic origin of
EOAD in the other members of this family.
Linkage of PSEN-2 mutations with the autosomal
transmission of familial EOAD has been shown for
13 different positions in the PSEN-2 gene (Table 1).
Estimations of the frequency of occurrence of muta-
tions in the PSEN-2 gene in pedigrees with suspected
familial EOAD vary between 3-5%. Whether there are
significant differences in clinical symptomatology be-
Table 1. Known mutations of the PSEN-2 gene on chromo-
some 1 associated with early-onset Alzheimer’s disease
(EOAD). http://www.molgen.ua.ac.be/ADMutations/
1 Arg 62 His 4 / N -Term Cruts et al. 1998
2 Ala 85 Val 4 / N -Term Piscopo et al. 2005
3 Thr 122 Pro 5 / HL - I Finckh et al. 2000
4 Thr 122 Arg 5 / HL - I Binetti et al. 2003
5 Ser 130 Leu 5 / HL - I Sorbi et al. 2002
6 Asn 141 Ile 5 / TM - II Levy-Lahad et al. 1995
7 Val 148 Ile 5 / TM - II Beyer et al. 1998
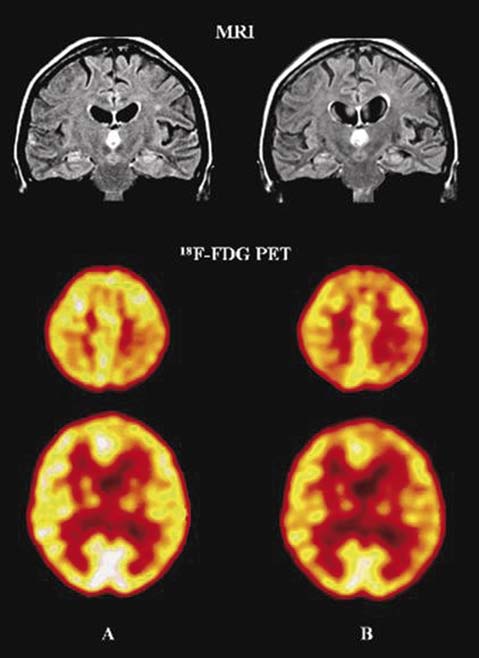
Fig. 2. MRI scans shown atrophy of the left entorhinal cor-
8 Gln 228 Leu 7 / TM - V Zekanowski et al. 2003
tex with relative sparing of the hippocampus.18F-FDG PET
scans showing progressive hypometabolism of both tem-
9 Met 239 Val 7 / TM - V Rogaev et al. 1995
poroparietal lobes and left frontal lobe between May 2005, al-
ready beginning in the left parietal cortex (A) and February
10 Met 239 Ile 7 / TM - V Finckh et al. 2000
11 Pro 334 Arg 10 / HL - VI b Lleo et al. 2002
12 Thr 430 Met 12 / TM - IX Lleo et al. 2002
Herein we document clinical and apperative findings
in a German patient with early-onset Alzheimer de-
13 Asp439Ala 12 / C - Term Lleo et al. 2001
mentia (EOAD) and with a mutation at codon 141 in
the PSEN-2 gene. The same mutation has been shown
tween different mutations of the PSEN-2 gene awaits
measurements of CSF-tau and CSF-Aß1-42 have been
further investigation. Some of these families show a
suggested to increase the diagnostic precision of AD
wide range in age at onset, and cases of nonpene-
(Galasko 1998; Hulstaert et al. 1999). As part of the
trance have been found (Cruts et al. 1998; Finckh et al.
clinical routine, these markers have been found to be
2005; Levy-Lahad et al. 1995; Tomaino et al. 2007).
highly sensitive and specific (Andreasen et al. 1998;
In our family, there was an almost 15-year gap be-
tween the proband and parent in age at disease onset.
Hampel et al. (2004) studied 52 patients with MCI,
Although a longer follow-up period is needed, the
93 AD patients, and 10 healthy controls (HC). The
presence of a cognitively healthy mutation carrier
MCI group was composed of 29 patients who had
could indicate the existence of incomplete penetrance.
converted to AD during follow-up, and of 23 patients
Nevertheless, we can not rule out that this individual is
who showed no cognitive decline. The levels of CSF
at risk for EOAD. The variation in age at onset ob-
tau-protein were increased, whereas levels of Aß1-42
served in this family confirms that PSEN2 mutations
were decreased in MCI subjects. Aß1-42 predicted AD
are associated with variable clinical expression. This
in converted MCI with a sensitivity of 59% and a
fact has important consequences for genetic-testing
specificity of 100% compared to HC. Tau-protein
and genetic-counseling programs; it may determine
yielded a greater sensitivity of 83% and a specificity of
the type of information given to these families.
The N141I mutation is located in exon 5 of the
Regarding its potential role in AD, studies with
PSEN2 gene, near the D439A mutation and the C-ter-
transfected cell lines and transgenic animals expressing
minal end of the protein. The presenilin 2 protein is
mutant PSEN-2 showed an alteration in amyloid pro-
an integral transmembrane protein normally processed
cessing leading to a higher production of Aß1-42 /1-
by proteolytic cleavage. Interestingly, the C-terminus is
43 (Citron et al. 1997; Tomita et al. 1997).
a critical region for endoproteolytic processing and
possibly for the pathologic function of the protein
demonstrated that the missense mutation at codon
(Shirontani et al. 2000) Thus; the N141I mutation
141 of the PSEN2 gene, the MCI present during the
could disrupt the endoproteolytic process and inter-
first consultation and the elevated tau-protein levels
fere with the normal function of the protein.
and decreased Aß1-42 levels in CSF interact in their
The diagnosis of primary degenerative dementia
effect on brain metabolism in specific brain areas. Our
disorders such as AD is made largely by excluding oth-
findings are interesting as several 18F-FDG PET stud-
er causes of dementia. The search for biochemical di-
ies have been published predicting the decline from
agnostic markers that could be used for an early diag-
normal to MCI. This studies showed that reduced
nosis of AD has led to the suggestion that the con-
baseline metabolic levels in the entorhinal cortex,
centrations of tau-protein and the 42-amino acid form
which is part of the medial temporal lobes (MTL, i.e.,
of Aß1-42 in cerebrospinal fluid (CSF) have a diag-
hippocampus, transentorhinal and entorhinal cortices,
nostic value (Andreasen et al. 2001; Hulstaert et al.
and parahippocampal gyrus), predict an MCI diagnosis
3 years later (de Leon et al. 2001; Gary et al. 2000) The
Tau-protein is a normal axonal protein, which by
baseline metabolic reduction predicted decline to MCI
binding to tubulin in microtubules promotes their as-
with 83% sensitivity and 85% specificity. No cortical
sembly and stability (Goedert 1993). An increase in
regions showed preclinical effects. Moreover, progres-
CSF-tau-protein in AD has been found in numerous
sive metabolic reductions in the entorhinal cortex and
studies (Arai et al. 1997; Blennow et al. 1995; Jensen et
in the left lateral temporal lobe paralleled the onset of
al. 1995; Vandermeeren et al. 1993), which probably
MCI, which is of interest, as an ideal biomarker for
reflects the neuronal and axonal degeneration
EOAD must correlate with disease progression.
(Blennow et al. 1995; Vanmechelen et al. 1996), or
possibly the successive accumulation of neurofibrillary
tangles in AD (Tapiola et al. 1997). The sensitivity of
CSF-tau-protein for AD in several studies has been
Although mutations in the genes for PSEN-1, PSEN-
high, often 80-90% (Andreasen et al. 1998, Andreasen
2 and APP cause familial EOAD, it must be remem-
et al. 1999; Galasko 1998). The specificity has also
bered that the majority of EOAD is not genetically
been relatively high because most patients with other
determined but belongs to the sporadic type of AD.
dementias, chronic neurologic disorders (e.g., Parkin-
EOAD due to a mutation in the PSEN-2 gene seems
son disease), or psychiatric diagnoses (e.g., depressive
to be rare in Germany. Our findings support the no-
pseudo-dementia) have physiologic CSF tau-protein
tion that CSF tau-protein, Aß1-42 and structural and
values (Blennow et al. 1995; Mecocci et al. 1998; Sjö-
functional neuroimanging (MRI, 18F-FDG PET) may
be useful biomarkers in the early identification of AD
Aß1-42 has been implicated in the pathogenesis of
AD and is the core peptide that accumulates in senile
plaques (Tamaoka et al. 1995). Several studies have
found that CSF- Aß1-42 is decreased in AD (An-
dreasen et al. 1999; Galasko et al. 1997; Motter et al.
1995; Sjögren et al. 2000). A high sensitivity (80-90%)
Acknowledgements: We thanks Ulrich Müller and Dagmar
for CSF- Aß1-42 as a marker for AD has been found
Nolte (Institute of Human Genetics at the University of
(Andreasen et al. 1999; Galasko 1998), whereas the
Gießen/Marburg, Germany) for the critical, stimulating and
specificity has to be investigated further. Concomitant
motivating comments on the final version of the article.
tients with Early-Onset Dementia Detected by Sequence
Analyses of Four Different Genes. American Journal of
Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A,
Winblad B. Cerebrospinal fluid beta-amyloid (1–42) in
Galasko D. CSF tau and Aß42: logical biomarkers for
Alzheimer disease: differences between early- and late-on-
Alzheimer’s disease? Neurobiol Aging 1998;19:117-9.
set Alzheimer disease and stability during the course of
Galasko D, Clark C, Chang L, Miller B, Green RC, Rotter R.
disease. Arch Neurol 1999;56:673-80.
Assessment of CSF levels of tau protein in mildly de-
Andreasen N, Minthon L, Davidsson P, Vanmechelen E,
mented patients with Alzheimer’s disease. Neurology
Vanderstichele H, Winblad B, Blennow K. Evaluation of
CSF-tau and CSF-Aß42 as diagnostic markers for
Gary W, Small L, Ercoli D. Cerebral metabolic and cognitive
Alzheimer’s disease in clinical practice. Arch Neurol
decline in persons at genetic risk for Alzheimer's disease.
Andreasen N, Vanmechelen E, Van de Voorde A, Davidsson
Goedert M. Tau protein and the neurofibrillary pathology of
P, Hesse C, Tarvonen S. Cerebrospinal fluid tau protein
Alzheimer’s disease. Trends Neurosci 1993;16:460-5.
as a biochemical marker for Alzheimer’s disease: a com-
Hampel H, Teipel SJ, Fuchsberger T, Andreasen N, Wiltfang
munity based follow up study. J Neurol Neurosurg Psy-
J, Otto M, Shen Y, Dodel R, Du Y, Farlow M, Möller H-
J, Blennow K, Buerger K. Value of CSF ß-amyloid 1–42
Arai H, Nakagawa T, Kosaka Y, Higuchi M, Matsui T, Oka-
and tau as predictors of Alzheimer’s disease in patients
mura N. Elevated cerebrospinal fluid tau protein level as
with mild cognitive impairment. Molecular Psychiatry
a predictor of dementia in memory-impaired individuals.
Hulstaert F, Blennow K, Ivanoiu A, Schoonderwaldt HC,
Arai H, Terajima M, Miura M, Higuchi S, Muramarsu T,
Riemenschneider M, De Deyn PP. Improved discrimina-
Machida N. Tau in cerebrospinal fluid: a potential diag-
tion of AD patients using ß-amyloid (1–42) and tau levels
nostic marker in Alzheimer’s disease. Ann Neurol
Jensen M, Basun H, Lannfelt L. Increased cerebrospinal fluid
Beyer K, Lao JI, Fernandández-Novoa L, Cacabelos R. Iden-
tau in patients with Alzheimer’s disease. Neurosci Lett
tification of a novel mutation (V148I) in the TM2 domain
of the presenilin 2 gene in a patient with late-onset
Lao JI, Beyer K, Novoa LF, Cacabelos R. A novel mutation
Alzheimer disease. Neurobiology of Aging 1998;19:87-8.
in the predicted TM2 domain of the presenilin 2 gene in a
Binetti G, Signorini S, Squitti R, Alberici A, Benussi L, Cas-
Spanish patient with late-onset disease. Neurogenetics
setta E, Frisoni GB, Barbiero L, Feudatari E, Nicosia F,
Testa C, Zanetti O, Gennarelli M, Perani D, Anchisi D,
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J,
Ghidoni R, Rossini PM. Atypical dementia associated
Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K,
with a novel presenilin-2 mutation. Annals of Neurology
Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E,
Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE.
Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Van-
Candidate gene for the chromosome 1 familial
mechelen E. Tau protein in cerebrospinal fluid: a bio-
Alzheimer's disease locus. Science 1995;269:973-7.
chemical marker for axonal degeneration in Alzheimer
Lleo A, Blesa R, Gendre J, Castellvi M, Pastor P, Queralt R,
disease? Mol Chem Neuropathol 1995;26:231-45.
Oliva R. A novel presenilin 2 gene mutation (D439A) in a
Citron M, Westaway D, Xia WM, Carlson G, Diehl T,
patient with early-onset Alzheimer's disease. Neurology
Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis
A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao
Lleo A, Blesa R, Queralt R, Ezquerra M, Molinuevo JL, Pena-
H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk
Casanova J, Rojo A, Oliva R. Frequency of mutations in
D, Fraser P, Hyslop PS, Selkoe DJ. Mutant presenilins of
the presenilin and amyloid precursor protein genes in ear-
Alzheimer’s disease increase production of 42-residue
ly-onset Alzheimer disease in Spain. Archives of Neurolo-
amyloid ß-protein in both transfected cells and transgenic
Mann DMA, Iwatsubo T, Nochlin D, Sumi SM, Levy-Lahad
Cruts M, van Duijn CM, Backhovens H, Van den Broeck M,
E, Bird TD. Amyloid (Ab) deposition in chromosome 1-
Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy
linked Alzheimer’s disease: the Volga German families.
J, St George- Hyslop PH, Hofman A, Van Broeckhoven
C. Estimation of the genetic contribution of presenilin-1
Mecocci P, Cherubini A, Bregnocchi M, Chionne F, Cecchetti
and -2 mutations in a population-based study of presenile
R, Lowenthal DT. Tau protein in cerebrospinal fluid: a
Alzheimer disease. Human Molecular Genetics 1998;7:43-
new diagnostic and prognostic marker in Alzheimer dis-
ease? Alzheimer Dis Assoc Disord 1998;12:211-4.
de Leon MJ, Convit A, Wolf OT. Prediction of cognitive de-
Morris JC, Price AL. Pathologic correlates of nondemented
cline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-
aging, mild cognitive impairment, and early-stage
Dglucose/positron-emission tomography (FDG/PET).
Alzheimer’s disease. J Mol Neurosci 2001;17:101-18.
Proc Natl Acad Sci 2001;98:10966-71.
Motter R, Vigo Pelfrey C, Kholodenko D, Granerus A-K,
Ezquerra M, Lleo A, Castellvi M, Queralt R, Santacruz P,
Clarberg A, Vanderstichele H. Reduction of ß-amyloid
Pastor P, Molinuevo JL, Blesa R, Oliva R. A novel muta-
peptide 42 in the cerebrospinal fluid of patients with
tion in the PSEN2 gene (T430M) associated with variable
Alzheimer’s disease. Ann Neurol 1995;38:643-8.
expression in a family with early-onset Alzheimer disease.
Mullan M, Crawford F. Genetic and molecular advances
Archives of Neurology 2003;60:1149-51.
in Alzheimer’s disease. Trends Neurosci 1993;16:398-
Finckh U, Kuschel C, Anagnostouli M, Patsouris E, Pantes
GV, Gatzonis S, Kapaki E, Davaki P, Lamszus K,
Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG,
Stavrou D, Gal A. Novel mutations and repeated findings
Kokmen E. Mild cognitive impairment: clinical character-
of mutations in familial Alzheimer disease. Neurogenetics
ization and outcome. Arch Neurol 1999;56:303-8.
Piscopo P, Crestini A, Malvezzi-Campeggi L, Manfredi A,
Finckh U, Muller-Thomsen T, Mann U, Eggers C, Markstein-
Deiana E, Cherchi R, Vanacore N, Marcon G, Piras MR,
er J, Meins W, Binetti G, Alberici A, Hock C, Nitsch RM,
Confaloni AM. A novel PSEN-2 mutation in a large Ital-
Gal A. High Prevalence of Pathogenic Mutations in Pa-
ian pedigree. Alzheimer's and Parkinson's Diseases: In-
sights, Progress and Perspectives. 7th International Con-
Tomaino C, Bernardi L, Anfossi M, Costanzo A, Ferrise F,
ference AD/PD Book of Abstracts 2005;24.
Gallo M, Geracitano S, Maletta R, Curcio SA, Mirabelli
Riemenschneider M, Buch K, Schmolke M, Kurz A, Guder
M, Colao R, Frangipane F, Puccio G, Calignano C, Mura-
WG. Cerebrospinal protein tau is elevated in early
ca MG, Paonessa A, Smirne N, Leotta A, Bruni AC. Pre-
Alzheimer’s disease. Neurosci Lett 1996;212:209-11.
senilin 2 Ser130Leu mutation in a case of late-onset "spo-
Riemenschneider M, Lautenschlager N, Wagenpfeil S, Diehl
radic" Alzheimer's disease. Journal of Neurology 2007;
J, Drzezga A, Kurz A. Cerebrospinal fluid tau and beta-
amyloid 42 proteins identify Alzheimer disease in subjects
Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K,
with mild cognitive impairment. Arch Neurol 2002;59:
Tokuhiro S, Capell A, Walter J, Grunberg J, Haass C,
Iwatsubo T, Obata K. The presenilin 2 mutation (N1411)
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda
linked to familial Alzheimer’s disease (Volga German
M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L,
families) increases the secretion of amyloid beta ending at
Sorbi S, Nacmias B, Piacentini S, Amaducci L, Chumakov
the 42nd (or 43rd) residue. Proc Natl Acad Sci USA
I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St
George-Hyslop PH. Familial Alzheimer's disease in kin-
Vandermeeren M, Mercken M, Vanmechelen E, Six J, van de
dreds with missense mutations in a gene on chromosome
Voorde A, Martin JJ, Cras P. Detection of tau proteins in
1 related to the Alzheimer's disease type 3 gene. Nature
normal and Alzheimer’s disease cerebrospinal fluid with a
sensitive sandwich enzyme-linked immunosorbent assay.
Rosenberg RN. The molecular and genetic basis of AD: the
end of the beginning. Neurology 2000;54: 2045-54.
Vanmechelen E, Blennow K, Davidsson P, Cras P, Van de
Sherrington R, Rogaev EI, Liang Y. Cloning of a gene bear-
Voorde A. Combination of tau/phospho-tau with other
ing missense mutations in early-onset familial Alzheimer’s
biochemical markers for Alzheimer CSF diagnosis and
tau in CSF as marker for neurodegeneration. In: Iqbal K,
Shirotani K, Takahashi K, Araki W. Mutational analysis of in-
Winblad B, Nishimura T, Takeda M, Wisniewski HM
trinsic regions of presenilin 2 that determine its endopro-
(eds) Alzheimer’s disease: biology, diagnosis and thera-
teolytic cleavage and pathological function. J Biol Chem
peutics. Wiley, Chicheste, pp. 1996;197-203.
Zekanowski C, Styczynska M, Peplonska B, Gabryelewicz T,
Sjögren M, Minthon L, Davidsson P, Granerus A-K, Clarberg
Religa D, Ilkowski J, Kijanowska-Haladyna B, Kotapka-
A, Vanderstichele H. CSF levels of tau, ß-amyloid 42 and
Minc S, Mikkelsen S, Pfeffer A, Barczak A, Luczywek E,
GAP-43 in frontotemporal dementia, other types of de-
Wasiak B, Chodakowska-Zebrowska M, Gustaw K,
mentia and normal aging. J Neural Transm 2000;105:563-
Laczkowski J, Sobow T, Kuznicki J, Barcikowska M. Mu-
tations in presenilin 1, presenilin 2 and amyloid precursor
Sorbi S, Tedde A, Nacmias B, Ciantelli M, Caffarra P, Ghi-
protein genes in patients with early-onset Alzheimer's dis-
doni E, Bracco L, Piccini C. Novel presenilin 1 and prese-
ease in Poland. Experimental Neurology 2003;184:991-6.
nilin 2 mutations in early-onset Alzheimer's disease fami-
lies. Neurobiology of Aging 2002;23:312-3. Received: June 4, 2008 / Accepted: July 21, 2008
Tamaoka A, Sawamura N, Odaka A, Suzuki N, Mizusawa H,
Shoji S. Amyloid ß protein 1–42/43 (Aß1–42/43) in cere-
bellar diffuse plaques: enzyme-linked immunosorbent as-
say and immunocytochemical study. Brain Res 1995;679:
Tapiola T, Overmyer M, Lehtovirta M, Helisalmi S, Ramberg
J, Alafuzoff I. The level of cerebrospinal fluid tau corre-
Department of Psychiatry and Psychotherapy
lates with neurofibrillary tangles in Alzheimer’s disease.
Tedde A, Nacmias B, Ciantelli M, Forleo P, Cellini E, Bagnoli
S, Piccini C, Caffarra P, Ghidoni E, Paganini M, Bracco
L, Sorbi S. Identification of new presenilin gene muta-
tions in early-onset familial Alzheimer disease. Archives
Metro Anesthesia & Pain Management NAME______________________________BIRTH DATE_____________AGE ________DATE___________________ REFERRING DOCTOR________________________________FAMILY DOCTOR_____________________________ Where is your pain? _________________________________________________________________________________ Does your pain radiate to anywhere? ______________________ _____
 to cosegregate in an autosomal dominant way with
EOAD in Volga-Germans in the U.S. (Levy-Lahad et
This case is unique in many respects. Although the
symptoms, signs, investigations, progress, and family
history are consistent with familial EOAD, the neu-
ropsychological follow-up changes of her affected rel-
atives – who shared a similar presentation – showed
the changes seen most often in MCI (Peterson et al.
to cosegregate in an autosomal dominant way with
EOAD in Volga-Germans in the U.S. (Levy-Lahad et
This case is unique in many respects. Although the
symptoms, signs, investigations, progress, and family
history are consistent with familial EOAD, the neu-
ropsychological follow-up changes of her affected rel-
atives – who shared a similar presentation – showed
the changes seen most often in MCI (Peterson et al.