Tadalafil entfaltet seine Wirkung über eine selektive Hemmung der PDE5, wodurch die Konzentration von cGMP im glatten Muskelgewebe stabil bleibt. Diese biochemische Modulation resultiert in einer langanhaltenden Relaxation der Gefäßwände. Der Wirkstoff wird nach oraler Einnahme effizient resorbiert, mit einer Bioverfügbarkeit von rund 80 %. Seine Halbwertszeit von bis zu 36 Stunden ist innerhalb dieser Substanzklasse außergewöhnlich. Abgebaut wird er in der Leber, hauptsächlich durch CYP3A4, mit anschließender biliärer Exkretion. Typische unerwünschte Wirkungen entstehen durch eine verstärkte Vasodilatation, etwa Kopfschmerzen oder Flush. Pharmakologisch wird cialis generika vor allem durch die verlängerte Wirkungsdauer charakterisiert.
Personnelhub.co.uk
The importance of testing for adrenoleucodystrophy inmales with idiopathic Addison’s disease
M D Ronghe, J Barton, P E Jardine, E C Crowne, M H Webster, M Armitage, J T Allen,C G Steward. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
X linked adrenoleucodystrophy (X-ALD) is considered to be a rare cause of Addison’s disease, although
several small series suggest a high incidence in young Addisonian males. A survey in the south west of
. . . . . . . . . . . . . . . . . . . . . . .
England identified 12 male patients diagnosed with Addison’s disease in the period 1987–99. In 10
of these (83%) X-ALD was the underlying cause; the other two were of autoimmune aetiology. Five boys
had developed Addison’s disease subsequent to the diagnosis of X-ALD. Of the remaining five, in three
boys the diagnosis of X-ALD was considerably delayed (by six months to two years from that of Addi-
Children, Upper MaudlinStreet, Bristol BS2 8BJ, UK;
son’s disease) and in two it was only made as a result of this survey. We also identified a patient who
presented with Addison’s disease at the age of 5 years but was only diagnosed as having X-ALD at theage of 34 years; in the interim his diagnosis of adrenomyeloneuropathy had been missed. Our experi-
ence highlights the absolute necessity of measuring very long chain fatty acids in all males with idio-
. . . . . . . . . . . . . . . . . . . . . . .
ThetermXlinkedadrenoleucodystrophy(X-ALD)denotes whenhedevelopedbehaviouralproblemsandschoolfailure
a group of diseases caused by defects in the ALD gene at
(patient A in table 2). We subsequently performed a search of
Xq28, which encodes a peroxisomal ATP binding cassette
the diagnostic index at the Royal Hospital for Sick Children,
transporter protein.1 Gene defects result in an extremely vari-
Bristol and sent a questionnaire to paediatricians and
able phenotype (see table 1), including a presymptomatic
endocrinologists throughout the south west of England
form, isolated adrenal insufficiency, devastating childhood
asking for notification of previous cases of Addison’s disease in
cerebral ALD (C-ALD), adult C-ALD, and adrenomyeloneu-
boys aged less than 16 years. Children with established
ropathy (AMN)—a disease causing spastic paraparesis and
aetiology (such as multiple endocrine deficiencies caused by
peripheral neuropathy in the second decade or beyond and
autoimmune disease or autoimmune polyendocrinopathy
leading to progressive impairment and early death. Despite
candidiasis ectodermal dystrophy) were excluded.
this array of subtypes and identification of at least 200 differ-
The diagnosis of primary adrenal failure was made on the
ent mutations, there appears to be no phenotype/genotype
basis of raised basal adrenocorticotrophic hormone (ACTH)
concentrations, together with a subnormal cortisol response
The tissues and body fluids of patients with X-ALD contain
to a standard Synacthen test. A standard dose of 250 µg of
abnormally high concentrations of unbranched saturated very
synthetic ACTH was administered and a peak cortisol value of
long chain fatty acids (VLCFA), particularly hexacosanoic acid
less than 500 nmol/l indicated adrenal insufficiency.
(C26:0) and tetracosanoic acid (C24:0). This excess is moststriking in the cholesterol ester and ganglioside fractions of
affected brain white matter and adrenal cortex, but is present
Serum cortisol concentrations were estimated using quantita-
to varying degrees in virtually all tissues and body fluids.1
tive sequential immunometric assay on the Immulite 2000
Diagnosis is made on the basis of raised concentrations of
analyser. Serum ACTH concentrations were measured on the
VLCFA in plasma and/or cultured skin fibroblasts.1–3
Nichols Advantage by a two site chemiluminescence immu-
Adrenocortical insufficiency is present in at least 50% of
noassay, utilising one mouse monoclonal antibody and a goat
patients with C-ALD/AMN, but may be the only clinical mani-
polyclonal antibody. Plasma renin activity was measured
festation of X-ALD in up to 10% of cases. As a consequence of
using radioimmunoassay following generation of angiotensin
the rarity of X-ALD (minimum incidence of approximately 1
I formed after incubation of plasma at 37°C for three hours in
in 20 000 white males1), this disease only accounts for a small
the presence of angiotensin converting enzyme inhibitor.
proportion of all cases of adrenal insufficiency. However, it has
Plasma concentrations of VLCFA were measured by
recently been recognised in high frequency in young males
capillary gas chromatography–mass spectrometry in the
with idiopathic Addison’s disease; representative series report
regional laboratory. The reference range for C26 was 0.33–1.39
5/8 boys,4 and 2/2,5 4/12,6 5/14,7 and 5/248 adolescent or adult
µmol/l; ranges for coefficients C26/C22 and C24/C22 were
0–0.03 and 0.32–0.90 respectively.
In order to both emphasise and extend these observations
we report our findings in the only 12 boys in whom Addison’s
disease has been diagnosed in the south west of England since
In addition to the index patient, our enquiry revealed 11 other
1987. Ten of these boys were affected by X-ALD.
boys with Addison’s disease and a 34 year old man who had
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
This study was prompted by a child who had presented with
Abbreviations: ACTH, adrenocorticotrophic hormone; ALD,
Addisonian crisis in response to pneumonia but whose under-
adrenoleucodystrophy; AMN, adrenomyeloneuropathy; C-ALD, cerebral
lying diagnosis of cerebral ALD was only made two years later
ALD; VLCFA, very long chain fatty acid; X-ALD, X linked ALD
Diminishes with ageCommon <4 yearsVery rare >40 years
presented to paediatric care with Addison’s disease at the age
disease to that of X-ALD ranged from 6 months to 29 years in
of 5 years. Of these 12 patients, none had idiopathic disease;
two had autoimmune disease proven by positive adrenal
Assay of VLCFA in plasma is the most frequently used test
autoantibodies, and the remaining 10 all had elevation of
for diagnosis of X-ALD and is accurate in almost all cases. No
VLCFA, diagnostic of underlying X-ALD.
false negative results were documented in the largest series
Table 2 gives clinical and biochemical data on the six
which included samples from more than 30 000 individuals
patients with X-ALD (including the index case) presenting
analysed at the Kennedy Krieger Institute.1 However, two false
with Addison’s disease; table 3 presents the remaining five
negative plasma VLCFA assays have been reported.2 3 In both
patients with X-ALD who went on to develop Addison’s
these cases the diagnosis of X-ALD was subsequently
disease later in their disease course.
confirmed by assays of VLCFA concentrations in cultured skin
In addition to a subnormal response to a standard
fibroblasts. Hence in patients with a high index of suspicion
Synacthen test, ACTH concentrations were raised in each boy
but normal or borderline plasma VLCFA, concentrations
studied (see tables 2 and 3), indicating primary adrenocortical
should also be measured in the cultured skin fibroblasts before
insufficiency. The reference range of ACTH used was 5–36 ng/l.
excluding the diagnosis of X-ALD, and a close interaction
In addition, renin activity was measured; the reference range
between clinicians and the laboratory is of utmost import-
Of the 10 patients identified by the survey, five had
The current study does not purport to be free of ascertain-
Addison’s disease as their initial presentation, but in only one
ment bias, perhaps the best example being inclusion of an
case was a prompt diagnosis of biochemical ALD made
adult patient originally diagnosed with Addison’s disease in
because of physician awareness (patient B). Of the remaining
1968. However, as a result of the comprehensive tertiary refer-
four boys, two were diagnosed only as a consequence of this
ral service maintained in South West England it is most
study, another because his mother requested a test on the
unlikely that any boys presenting with Addison’s disease in
basis of a previous family history (when her son became Add-
the past decade would have been missed. The finding that 11
isonian at 9 years of age), and the fourth 29 years after his ini-
of 13 paediatric presentations of Addison’s disease were
tial presentation with Addison’s disease. This last patient had
caused by underlying X-ALD is also consistent with the only
undergone bladder neck excision for presumed outflow tract
previous paediatric series, where 5/8 boys with Addison’s dis-
obstruction, but his underlying diagnosis of AMN was only
appreciated when he developed paresthesiae and gait prob-
Although accurate diagnosis of X-ALD does not affect the
lems during convalescence from surgery.
management of endocrine problems in the index case, it opens
The remaining five had developed Addison’s disease after a
the possibility of therapy before the onset of overt neurologi-
primary diagnosis of X-ALD. Two of these five developed
cal disease. Neurological manifestations of X-ALD range from
Addison’s disease subsequent to an initial diagnosis of severe
devastating leucoencephalopathy (fig 1 shows a magnetic
C-ALD (one had a previous family history of C-ALD in a cousin
resonance imaging (MRI) scan in the index case) within the
and could have been screened three years previously, although
first decade to spastic paraparesis or psychiatric presentations
this had not been offered). The remaining three had a diagno-
in later adult life; all may occur within the same family. It is
sis of biochemical ALD (raised VLCFA but no other indicator of
thought that neurological disease will develop in up to 90% of
disease); these patients had all been diagnosed because of a
patients with the biochemical defect of X-ALD. In the current
preceding family history of ALD/AMN. In these five patients,
series, although the median follow up is only 5 years, only four
the interval between the diagnosis of biochemical ALD or
of the 11 males remain completely free of radiological or clini-
C-ALD and the onset of adrenocortical insufficiency ranged
cal signs of this disease. It must be remembered that the delay
from six months to five years (see table 3).
from onset of adrenal insufficiency to the development ofneurological disability is highly variable and may be as long as32 years.12 This is exemplified by patient F in whom Addison’s
disease was diagnosed almost 30 years before developing
X-ALD is described as a “rare” cause of Addison’s disease in
the current editions of the Oxford textbook of medicine and Harri-
Current therapeutic choices lie between dietary therapy
son’s principles of internal medicine, and an “uncommon
(including Lorenzo’s Oil) for those with presymptomatic
association” in Forfar and Arneil’s textbook of paediatrics.9–11 This is
disease and bone marrow transplantation for those with early
largely a consequence of the majority of cases being females
with autoimmune disease or patients with tuberculosis. How-
C-ALD.12–15 Benefit from the latter appears to rely on slow
ever, in the small subset of patients who are young and male,
replacement of CNS microglial cells with cells of donor origin,
we believe these statements to be dangerously misleading and
since these arise from the bone marrow. As the childhood cer-
likely to result in recurrent late diagnosis of X-ALD. This com-
ebral form of this disease usually progresses rapidly over 6–18
promises the management of both patients and their extended
months, it is imperative to identify patients at a presympto-
families. In this series, delay from the diagnosis of Addison’s
matic stage. Careful monitoring can then be conducted,
Summary of clinical and laboratory data in patients with X-ALD presenting with Addison’s disease
uncle who developedAddison’s disease at11 y, AMN at 39 y,died at 42 y)
obstruction,parasthaesia and gaitproblems
*Age at diagnosis of Addison’s disease. FTT, failure to thrive.
Reference ranges: ACTH, 5–36 ng/l; renin, 0.5–3.1 pmol/ml/h; C26, 0.33–1.39 µmol/l.
MRI scan of brain, showing diffuse high signal
abnormalities in the periventricular white matter of both parietal and
occipital lobes (marked by arrows) in an advanced case of
allowing patients who are developing cerebral ALD to be
referred for bone marrow transplantation before overt neuro-
logical involvement. More recently lovastatin and sodiumphenylacetate have been shown to lower VLCFA in cultured
skin fibroblasts, although their therapeutic potential remainsunclear.16
Of equal importance to these therapeutic considerations are
the implications for the patient’s extended family. The identi-
fication of X-ALD in a family allows screening of other malerelatives who may be at risk for developing Addison’s disease
or neurological complications. In addition to providing the
option of early treatment, this may avoid deaths in affected
males—at least 10 patients are known to have died of Addiso-
nian crisis secondary to X-ALD, some of whom were free of
neurological disease.1 Similarly, carrier females may develop
milder neurological problems such as spastic paraparesis
(although curiously Addison’s disease is extremely rare in
Most importantly, early diagnosis brings the possibility of
genetic counselling, carrier detection, and antenatal diagno-
sis, and so the potential for radically reducing the incidence of
this devastating disease. For this reason above all, assessment
of plasma VLCFA concentrations should be regarded as man-
datory in the investigation of males with unexplained
We gratefully acknowledge the support of the COGENT Trust; Drs
Julian Pleydell-Pearce and Sunil Pullaperuma, who kindly provided
patient details; and Drs Charles Pennock and Janet Stone for manyhelpful biochemical discussions.
Adrenoleucodystrophy in Addison’s disease
. . . . . . . . . . . . . . . . . . . . .
6 Schafer JR, Ehlenz K, Steinmetz A, et al. Adrenomyeloneuropathy. A
frequent cause of Addison’s disease. Dtsch Med Wochenschr
M D Ronghe, C G Steward, Department of Haematology/Oncology,
7 Laureti S, Casucci G, Santeusanio F, et al. X-linked
Royal Hospital for Children, Upper Maudlin Street, Bristol BS2 8BJ, UK
adrenoleukodystrophy is a frequent cause of idiopathic Addison’s
P E Jardine, Department of Neurology, Royal Hospital for Children
disease in young adult male patients. J Clin Endocrinol Metab
E C Crowne, Department of Endocrinology, Royal Hospital for Children
J Barton, Department of Paediatrics, Royal Gwent Hospital, Cardiff
8 Jorge P, Quelhas D, Oliveira P, et al. X-linked adrenoleukodystrophy in
patients with idiopathic Addison’s disease. Eur J Pediatr
M H Webster, Department of Paediatrics, Taunton and Somerset
Hospital, Musgrove Park, Taunton TA1 5DA, UK
9 Edwards RW. Adrenocortical diseases. In: Weatherall DJ, Ledingham
JGG, Warrell DA, eds. Oxford textbook of medicine. Oxford: Oxford
M Armitage, Department of Endocrinology, The Royal Bournemouth
Medical Publications, 1996:1652–3.
Hospital, Castle Lane East, Bournemouth BH7 7DW, UK
10 William GH, Dluhy RG. Diseases of adrenal cortex. In: Braunwald E,
J T Allen, Biochemical Genetics Unit, The Lewis Laboratory, Clinical
Fauci AS, Kasper DL, et al, eds. Harrison’s principles of internal
Chemistry, Southmead Hospital, Westbury-on-Trym, Bristol BS10 5NB,
medicine. New York: McGraw-Hill, 2001:2098.
11 Kelner CJH. Endocrine gland disorders. In: Campbell AGM, McIntosh
N, eds. Forfar and Arneil’s textbook of paediatrics. Edinburgh: Churchill
12 Moser HW, Bergin A, Naidu S, Ladenson PW. Adrenoleukodystrophy.
1 Moser HW, Smith KD, Watkins PA, et al. X-linked
Endocrinol Metab Clin North Am 1991;20:297–318.
adrenoleukodystrophy. In: Scriver CR, Beaudet AL, Valle D, et al, eds.
13 Aubourg P, Blanche S, Jambaque I, et al. Reversal of early neurologic
The metabolic and molecular bases of inherited disease. New York:
and neuroradiologic manifestations of X-linked adrenoleukodystrophy by
bone marrow transplantation. N Engl J Med 1990;322:1860–6.
2 Kennedy CR, Allen JT, Fensom AH, et al. X-linked adrenoleukodystrophy
14 Krivit W, Lockman LA, Watkins PA, et al. The future for treatment by
with non-diagnostic plasma very long chain fatty acids. J Neurol
bone marrow transplantation for adrenoleukodystrophy, metachromatic
Neurosurg Psychiatry 1994;57:759–61.
leukodystrophy, globoid cell leukodystrophy and Hurlers syndrome. J
3 Wanders RJ, van Roermund CW, Lageweg W, et al. X-linked
Inherit Metab Dis 1995;18:398–412.
adrenoleukodystrophy: biochemical diagnosis and enzyme defect. J
15 Shapiro E, Krivit W, Lockman L, et al. Long term effect of bone marrow
transplantation for childhood onset cerebral X-linked
4 Sadeghi-Nejad A, Senior B. Adrenomyeloneuropathy presenting as
adrenoleukodystrophy. Lancet 2000;356:713–18.
Addison’s disease in childhood. N Engl J Med 1990;322:13–16.
16 Singh I, Pahan K, Khan M. Lovastatin and sodium phenylacetate
5 Kong M-F, Jeffcote W. Eighty-six cases of Addison’s disease. Clin
normalise the levels of very long chain fatty acids in skin fibroblasts of
X-adrenoleukodystrophy. FEBS Lett 1998;426:342–6.
Obesity and asthma are linked . . . really
Toavoiddevelopingasthmafatchildrenshouldloseweight.Alargecrosssectionalstudyofchildren
in the United States confirms that obesity increases the likelihood of asthma, but not atopy, as anindependent risk factor.
Previous studies linking body mass index (BMI) and respiratory symptoms including asthma have
been called into question by the recent finding that breastfeeding, which protects against asthma andatopy, is strongly inversely related to BMI in children starting school and is therefore a potential
Von Mutius et al sought to determine whether the apparent association between BMI and asthma is a
true one by analysing data on more than 7000 children from the National Health and Nutrition Exam-
ination Study (NHANES) III and data collected for a comprehensive range of variables. Among these were
prevalence of asthma and atopy and a whole array of known or potential confounders including birth
weight (children aged <12 years) and breastfeeding (children aged <6 years).
Prevalence of asthma and atopy increased significantly across the quartiles of BMI. The relation held
true for asthma after adjustment for potential confounders—age, sex, ethnicity, household size, and pas-sive exposure to smoke—and after further controlling for breastfeeding and birth weight. No independ-ent relation was seen between BMI and atopy. As cause and effect cannot be determined by a cross sec-tional study, the authors point to evidence from studies over time in nurses and children in support oftheir conclusion—that obesity predisposes to asthma. m Thorax 2001;56:835–838.
Knockoff: The Deadly Trade in Counterfeit Goods By Tim Phillips London, England & Sterling, VA; Kogan Page Ltd., 2005, ISBN 0-7494-4389-0 (Price $29.95), pp. 231 Reviewed by Erika Jacobsen White Journal of High Technology Law Suffolk University Law School The global counterfeit market currently wields nearly $538 billion annually. The U.S. counterfeit market alone is estimated to rake in
SpermMar IgA Test This can be done by means of the 10 microlitres capillary immunoglobulin class of antispermatozoal antibodies in serum. Am J Reprod Immunol Microbiol, 1985, 7 : 143-147. Test for determination of Sperm Antibodies Note: To use the microcapillary pipettes, insert the end of 4. FRIBERG J: Immunoglobulin concentration in serum and seminal fluid from men with and without
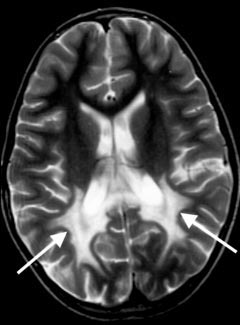 MRI scan of brain, showing diffuse high signal
abnormalities in the periventricular white matter of both parietal and
occipital lobes (marked by arrows) in an advanced case of
allowing patients who are developing cerebral ALD to be
referred for bone marrow transplantation before overt neuro-
logical involvement. More recently lovastatin and sodiumphenylacetate have been shown to lower VLCFA in cultured
skin fibroblasts, although their therapeutic potential remainsunclear.16
Of equal importance to these therapeutic considerations are
the implications for the patient’s extended family. The identi-
fication of X-ALD in a family allows screening of other malerelatives who may be at risk for developing Addison’s disease
or neurological complications. In addition to providing the
option of early treatment, this may avoid deaths in affected
males—at least 10 patients are known to have died of Addiso-
nian crisis secondary to X-ALD, some of whom were free of
neurological disease.1 Similarly, carrier females may develop
milder neurological problems such as spastic paraparesis
(although curiously Addison’s disease is extremely rare in
Most importantly, early diagnosis brings the possibility of
genetic counselling, carrier detection, and antenatal diagno-
sis, and so the potential for radically reducing the incidence of
this devastating disease. For this reason above all, assessment
of plasma VLCFA concentrations should be regarded as man-
datory in the investigation of males with unexplained
We gratefully acknowledge the support of the COGENT Trust; Drs
Julian Pleydell-Pearce and Sunil Pullaperuma, who kindly provided
patient details; and Drs Charles Pennock and Janet Stone for manyhelpful biochemical discussions.
MRI scan of brain, showing diffuse high signal
abnormalities in the periventricular white matter of both parietal and
occipital lobes (marked by arrows) in an advanced case of
allowing patients who are developing cerebral ALD to be
referred for bone marrow transplantation before overt neuro-
logical involvement. More recently lovastatin and sodiumphenylacetate have been shown to lower VLCFA in cultured
skin fibroblasts, although their therapeutic potential remainsunclear.16
Of equal importance to these therapeutic considerations are
the implications for the patient’s extended family. The identi-
fication of X-ALD in a family allows screening of other malerelatives who may be at risk for developing Addison’s disease
or neurological complications. In addition to providing the
option of early treatment, this may avoid deaths in affected
males—at least 10 patients are known to have died of Addiso-
nian crisis secondary to X-ALD, some of whom were free of
neurological disease.1 Similarly, carrier females may develop
milder neurological problems such as spastic paraparesis
(although curiously Addison’s disease is extremely rare in
Most importantly, early diagnosis brings the possibility of
genetic counselling, carrier detection, and antenatal diagno-
sis, and so the potential for radically reducing the incidence of
this devastating disease. For this reason above all, assessment
of plasma VLCFA concentrations should be regarded as man-
datory in the investigation of males with unexplained
We gratefully acknowledge the support of the COGENT Trust; Drs
Julian Pleydell-Pearce and Sunil Pullaperuma, who kindly provided
patient details; and Drs Charles Pennock and Janet Stone for manyhelpful biochemical discussions.